 学校主页
学校主页
 管理入口
管理入口

 发布于 2024-08-30
发布于 2024-08-30
 浏览量:
浏览量:近日,山东理工大学生命与医药学院姜立波教授、杨凤堂教授课题组与英国桑格研究所宁泽民教授团队合作,在生物信息学顶级期刊《Genomics, Proteomics & Bioinformatics》(IF=11.5)杂志上发表了一篇题为“The Bioinformatic Applications of Hi-C and Linked Reads”的文章。该论文的通讯作者为宁泽民教授,姜立波教授为第一作者。

在文中,研究团队重点探讨了短读长和长读长的long-range测序技术,这些技术能够提供更为丰富和全面的基因组信息,极大地促进了复杂基因组的组装和结构变异的挖掘。目前,有多种新兴测序技术能够生成高通量的long-range数据集,其中最具代表性的是Hi-C和Linked Reads技术。文章详细介绍了五种主要的long-range测序技术,包括Hi-C、10x Genomics Linked Reads、haplotagging、TELL-seq和stLFR的原理(图1),并探讨了它们的应用场景。研究团队还评估了不同技术所产生的测序数据质量,且总结了适用于long-range数据分析的生物信息学工具,为相关技术的使用提供了有价值的指导和借鉴。
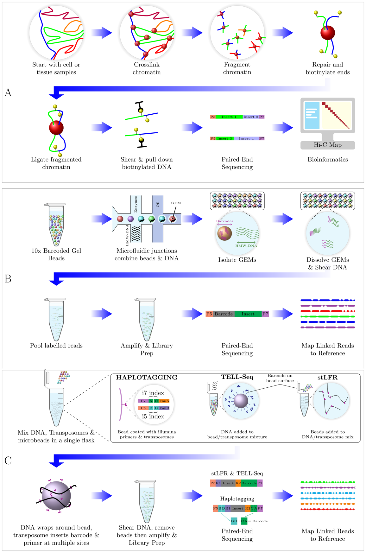
图1 五种long-range技术的文库构建的流程
Hi-C技术已经被广泛应用于基因组的scaffolding,通过使用不同的算法在染色体水平上组装基因组。相比之下,各种Linked Reads技术已被证明在提升基因组组装过程中short reads的价值方面具有显著优势。为此,文章提出了一些量化指标用于评估不同long-range技术平台的测序数据质量。研究结果表明,Hi-C Arima Genomics V2平台产生的数据质量明显优于V1平台 (图2)。每种技术都有其独特的特点,研究人员可以根据本文提出的评估方法来选择最适合的技术。总体而言,Hi-C和Linked Reads技术能够显著提高基因组组装质量,增强结构变异(SV)的检测精度。随着NGS技术和相关软件流程的进一步发展,long-range技术必将继续壮大。
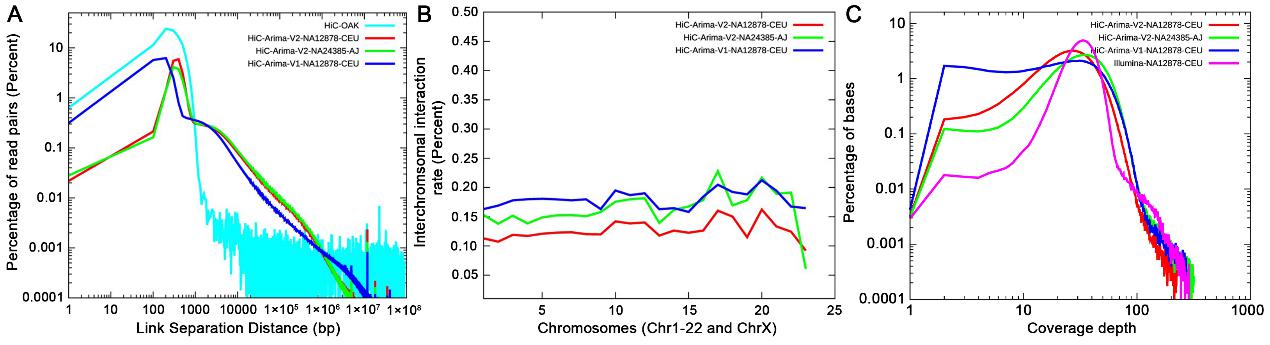
图2 Hi-C reads的特征
与long-read平台相比,long-range技术通过分割基因组和增加条形码来将reads聚类成组,从而获得所需的long-range信息。尽管其使用成本较标准的short-read测序略有增加,但其优势却相当明显。文章进一步证明,基因组的非局部信息能够有效补充long-read技术,并弥补了这些技术的一些缺陷。结合long-read和long-range方法,依然是研究复杂基因组、泛基因组组装、群体遗传学以及复杂性状高分辨率分析的首选策略。
目前,虽然Linked Reads已不再用于基因组组装,但其优势在于能够在低覆盖率下,以较低成本构建长单倍型,这使其非常适合大规模样本的研究。而且,Linked Reads的DNA输入要求低至1.5 ng,并且覆盖的均匀性较好。尽管long-read平台也在开发低输入模式的应用,例如PacBio的低输入和超低输入模式,但其最低要求仍为5 ng,且基本覆盖的均匀性不如普通文库。总体而言,Linked Reads仍然具有广阔的应用前景。
本文重点提出了数据质量控制的标准和方法,同时还提供了Hi-C和Linked Reads数据供读者练习。本研究得到了国家自然科学基金面上项目、山东省自然科学基金优秀青年基金以及英国惠康基金的资助支持。